lludin Sの構造、炭素番号、及び実験13C化学シフト値を以下に示しました

化学シフトを計算する。(Illudin Sを例として)
NMRで構造を決定するとき、自身の帰属にどうしても不安が残る時があります。
これを検証するとき、どのようにしていますか?
NMRの解析は、奥が深く経験を積むほど測定条件を含めて細かな議論が可能になります。
でも、構造的に新規な化合物の場合、経験的に積み上げてきた知見がなかなか使えない場合もあります。
神にもすがる思いで、根拠となるものを探そうとしますが、大きく分けて化学シフトの予想では演繹的手法と帰納的手法があります。
通常の解析も帰納的解析で、過去のデータが頭の中に残っていてこれを参照して構造を議論しています。
さらに、ウッドワード・フィーザー則のように、近似式に基づいて計算する手法も最近は利用可能です。
I官能基の数と分子サイズを考慮してlludin Sの計算を例に考えて見ます。
ここでは6-epi体を帰属してしまったと仮定します。
化学シフトは笠原先生の論文(食衛誌 Vol. 50, No. 4)に掲載されているデータを参考にしました(結構の炭素で帰属が間違っていました)。
lludin Sの構造、炭素番号、及び実験13C化学シフト値を以下に示しました
Chemdrawの機能を活用する
まずは、帰納的手法です。化学シフトの予想という点では、ChemDraw Professional版に付属するものを利用した方は多いのではないでしょうか
ChemNMRというデータベースソフトを参照して化学シフトを予想します。
この方法は構造式に基づき、データベースから適切な経験式を見出し、それにより化学シフトを予想しようというものです。
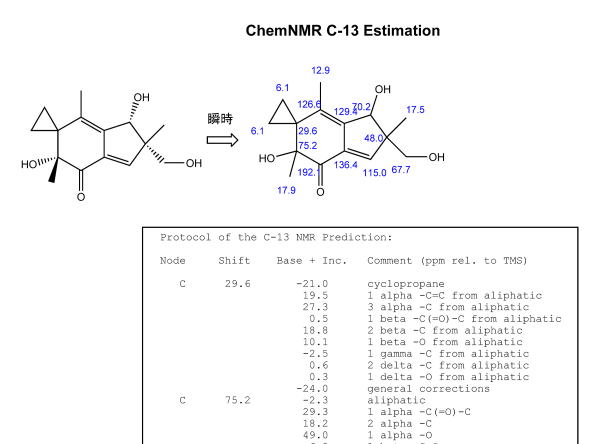
この方法は、ほぼ瞬時に1Hおよび13C化学シフトを計算してくれます。
Chemdrawで構造式を書いて、輪投げでくくって、「predict 13C chemical shifts」で完了、瞬時に別のファイルが現れ、構造上に化学シフト、また予想NMRチャート、計算の根拠などが示されます。
実験値との差異に対する二乗平均平方根(RMS値)は7.0 ppmでした。そんなに悪くないですよね。
自分の帰属に大きな間違いかないかをチェックするには十分な性能と言えます。
しかし、良く見るとこの方法では立体化学の概念が無視されていることがわかります。
ます。上の図ではシクロプロパンの二つのメチレン炭素の化学シフトは、実際には異なっていますよね。
したがって、この方法では、立体化学の違いを議論することが出来ないことがわかります。
現在、間違った6-epi構造を議論したいのですが、議論の根拠が有りません。
理論計算を行う。
ここで議論する化学シフトの理論計算とは、NMRの原理に基づき化学シフトを演繹的に求めようというものです。
具体的には各原子核の電子雲による遮蔽の度合いを分子軌道法に基づいて計算しようというものです。
したがって、電子の挙動の計算が重要となり、分子軌道計算が必要となります。
分子軌道計算には
1.Hartree-Fock近似式により波動関数を求める方法(HF法)
2.Kohn-Sham方程式により電子密度を求める方法(密度汎関数法、DFT法)
の二つがあります。構造そのものを議論するのであればどちらでも良いようですが、スペクトルなど励起状態が関与した物性を議論するにはDFT法の方が良いみたいです。
DFT法ではHF法と比較しても計算量が増大します。
したがって、少し前までは、DFT法による化学シフトの理論計算は、少し前までは大型計算機が必要でした。
コンピュータテクノロジーの目覚しい発展により、現在ではデスクトップパソコンですらそれが可能になってきました。
以下は、実際に使用している環境での説明になります。
パソコン: Core i7-6700 (3.4 GHz, 4コア、8スレッド)
Windows 7 (64 bit)
62GBメモリ
Spartanでは、計算の途中データをハードディスクに書き込むため、SSDを用いたほうが圧倒的に早くなるそうです。
optionでscratch spaceとして指定できるのでシステムディスクがSSDである必要はないそうです。
① 配座解析
化学シフトは、安定配座について計算を行う必要があります。
まず、illudin Sの構造をプログラム上で組み立て、配座解析の際、回転させる結合、環状構造ではフリップさせる炭素を指定します。
Spartanは回転・フリップさせる位置を自動的に指定する機能がありますが、そのままでは重要な配座を逃す可能性があります。
したがって、この指定は慎重に行う必要があります。
下の図は、デフォルトでは162配座の検索でしたが、カルボニル炭素フリップを追加して、324配座を検索するように指定しました。
計算は数秒で完了しました。
Spartanのデフォルトセッティングでは、最安定配座から40kJ/mol以内の配座かつ最大100配座がエネルギーの順でソートされた後、保存されます。
Illudinですと、配座の探索範囲は300配座前後と全く気になりませんが、回転を一箇所追加すると3倍、フリップは一箇所に付き2倍と探索範囲が広がります。
つまり、ちょっと複雑な化合物ですと数万配座を探索することになり大変です。
したがって、配座解析では安定配座の候補を求める程度に考えます。
得られた結果は、一つ一つ、分子をクルクル回して解析する必要があります。
どの段階でもいえるのですが、重要な配座が漏れている場合もあるので、完全な自動化は個人的には危険であると感じています。
配座検索ミスの理由:本当のことは良く判りませんが、個人的には歪みのある分子で多く遭遇する気がします。MMFF法などの力場パラメータは安定な分子を基に作成されており、安定構造を求める目的から最小値エネルギー付近のカーブ再現を重視していて、高エネルギー側については頓着できていない。或いはパラメータとして再現するほど実測値が無いのではと考えています。ひずみ分子の配座解析を行う場合は、計算に時間がかかるものの半経験分子軌道法やSTO3Gのようなコストの低い分子軌道法を用いるのも対応策の一つと考えています。
ただし、Spartan16から配座解析はデフォルトでは力場法のみとなってしまいました。
Spartan16でAM1を使った配座解析を行う場合:キーワードでAM1を入力することで行うことが出来ます。さらにキーワードに"SINGLESTEP "を追加する必要があります。
実際、illudin SでMMFFを使って配座探索した場合18安定配座が見つかり、AM1で探索した場合多は同じ設定で26安定配座が見つかりました。今回は最安定配座付近ではあまり影響はありませんでしたが、ひずみを持ったepoxyroussoeoneの場合は、配座漏れに悩まされました。epoxyroussoeoneの構造を計算で検証したメルク社のBuevich, Alexei V.からは、計算結果が違うとメールまで届きました。彼にはこちらの安定配座を送ることで納得してもらいました。彼の論文で、構造に間違いがないという結論をみてホッとした経験もあります。
今回は、34個の安定配座候補が見つかりました。
② 配座の絞込み
次により精密な手法により構造の精密安定化を行います。
計算の精度と所要時間はトレードオフの関係です。
力場計算により配座探索を行った直後は数十の配座が候補として保存されています。
これらの配座を全て高い精度で最適化しようとすると、非現実的な時間が要求されます。
そこで配座の絞込みを行います。
どの方法を使って使って絞り込むかの判断は難しいと思います。
そこで、illudin Sについて、MMFF力場で配座探索で見つかった34個の配座について、さまざまな方法で最適化したのが下のチャートです。
(近年のパソコンではセスキテルペン程度であれば、密度汎関数法で50配座程度なら最適化可能ですが、分子サイズが大きくなると非現実的です。)
半経験的分子軌道法はいずれも、安定配座候補の探索には有効でも、配座の絞込みには全く不適切であることも解ります。
このチャートを見る限り、STO3Gで得られる構造とNMR解析に用いるωB97X-D/6-31G*とは、配座の安定順位に大きな入れ替わりがなく、STO3Gでエネルギーウィンドウを多少大きく取れば大丈夫のように思えます。
今回の計算の場合、最安定配座から20KJ/mol以内に9配座となりました。
実際には、10kJ/molのエネルギー差があるとその配座存在比は98:2程度になってしまいます。
今回の計算では、最安定配座から10kJ/mnol以内には二つの配座しか存在しないことになります。
なお、赤矢印の配座のエネルギーのエネルギー順位が、大きく変化しています。
これは、構造最適化の際に、水酸基の立体配座が極大値を超えて隣の配座に移動してしまったためです。
隣の配座へのエネルギー障壁が大きくないと、時々このようなことが起きる様な気がしています。
これが、重要な配座漏れの原因の一つになっています。
このようなことから、計算結果については一つ一つ化学者の目でチェックする必要があります。
チェックしたところ、第二安定配座は3配座重複していました。分子の対称性が絡むような特殊な場合を除き、重複配座は削除する必要があります。
Spartan開発者のWarren Hehreによれば、配座の絞込みを自動化する機能をSpartanに組み込み、次期バージョンから利用可能にするとのことです。
なお、計算方法を変更した時ですが、spartan形式で保存すると、それ以前の計算履歴が残るようでファイルサイズが徐々に大きくなります。
新たな計算を行うと、"reading previsous wavewfunction”みたいな表示がなされるので、計算の効率化がなされるものと思います。
しかし観察してみるとは、構造最適化の勾配がゆるくなるのか、実際は計算時間は短縮されないどころか増大する傾向にあります。
一度、mol2形式で保存することで過去の計算履歴を完全に消去してから、再度spartanファイルを作成するとこれが回避されますが、果たして効果的なのかは自信がありません。
ちなみに、今回のilludin Sの配座絞込みでは1配座あたり以下の計算所要時間でした。(1CPUに換算しています。)
今回の結果ではSTO3GよりHF/3-21Gの方が計算時間が短かかったようです。(先ほどの計算履歴の消去と関連しているのかもしれません。)
計算方法 1配座あたりの所要時間(1CPU換算) MMFF 瞬時 PM3 3-5秒 AM1 1-3秒 RM1 1-3秒 STO3G 5-15分 HF/321G 5-15分 HF/631G* 30-90分 EDF2/6-31G* 90-120分 wB97X-D/6-31G* 4時間-6時間
③ 化学シフトの計算
化学シフトを計算します。
操作はいたって単純でNMR化学シフトのラジオボタンをクリックしてsubmitします。
既に、構造最適化が終了していますが、時には数回構造最適化をする場合もあります。
用いる汎関数によって結果は大きく左右されます。
どの汎関数を用いるかの選択はきわめて重要ですが、Spartan開発者のWarren HehreはωB97X-D/6-31G*の使用(Spartan16の場合)を推奨しています。
Spartan14の場合はEDF2/6-31G*でした。Spartan14ではωB97X-D/6-31G*の検証(補正)が行われておらず、計算は出来ますが、信頼性はまたく有りません。
実際には、構造変化は無視できるものなので計算時に構造最適化"equilibrium geometry"ではなく一点計算"energy"を選択して全く問題がないはずです。
ログでは並列処理を行っていると出てきますが、Spartan16の場合、1分子あたり5スレッドと指定しても、モニターではCPU timeがWall timeの2倍程度にしかなりませんので、並列処理の効率があまりよくないのかもしれません。
CPU負荷をモニターすると、負荷率は12%前後で推移しました。
8スレッド可能なシステムですので、多くの時間1スレッドしか使われていないようにも見えますが、設定どおりとはいえないまでも並列処理の効率は確かにあります。
今回の計算では、1配座あたり約30分(1CPUに換算)でした。
計算が終わると、各配座ごとに化学シフトを構造式上に示すことが出来ます(下図)。
計算では真空条件の配座を想定していますが、ベンゼンのような低極性溶媒の方が再現性が高いように感じています.
炭素核は水素原子にカバーされており、溶媒からのアニソトロピーは少ないはずです。
逆に、1H化学シフトの場合、溶媒のアニソトロピーの影響を受けやすいので、解析は多少難しいと感じています。
実際に1Hシグナルを比較しようとすると、分裂が多かったり、他のシグナルと重なってはっきりした化学シフトを求めにくい場合があり話が複雑になる場合が有ります。
実際には複数の配座の寄与があり、それらを配座の存在率によって重さをのせて平均を取ります。
Spartanには、その機能が充実しており以下のようなスプレッドシートを簡単に作成できます。
一番下の行には、ボルツマン分布を考慮した化学シフトとして表示されます。
④ データ解析
得られた計算結果を、統計的に処理します。
一般的に用いられるのは、実験値との差異を二乗平均平方根(root mean square, RMS)を求める方法です。
必要な式を作成してSpartanスプレッドシートで解析することも可能ですが、私はエクセルにデータを移して解析します。
スプレッドシートのLabelのところをクリックした後に右ボタンからコピーすることが出来ます。
実験値も入れて整理したものが下の表になります。
炭素番号 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 実験値(ppm) 200.6 76.2 30.8 135.1 138.4 74.9 55.0 141.4 136.0 24.7 6.0 8.7 14.2 15.8 69.0 Chemdrawの予想化学シフト
(ppm)191.2 75.2 29.6 126.6 129.4 70.2 48.0 115.0 136.4 17.9 6.1 6.1 12.9 17.5 67.7 実験値との差異 (ppm) 9.4 1.0 1.2 8.5 9.0 4.7 7.0 26.4 -0.4 6.8 -0.1 2.6 1.3 -1.7 1.3
ωB97X-D/6-31G*で求めた化学シフト配座ID 相対エネルギー (kJ/mol) ボルツマン分布 (%) 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 M0001 0.0 0.9 204.69 76.92 31.39 137.60 139.25 81.08 54.17 141.07 139.73 24.19 6.60 9.73 13.34 20.80 67.96 M0008 7.6 0.0 203.35 76.27 31.73 140.43 139.56 81.18 55.19 145.72 135.40 24.61 7.08 10.61 14.92 23.49 66.21 M0002 12.7 0.0 204.08 76.54 31.25 136.51 140.13 80.74 53.50 143.68 137.74 24.65 6.54 9.92 13.71 22.60 68.83 M0007 14.9 0.0 202.49 76.09 31.79 138.18 142.90 80.79 55.65 152.57 134.50 24.76 7.29 10.62 15.35 23.05 67.07 M0003 16.65 0.001 204.94 76.72 31.84 139.54 139.10 79.41 54.00 140.53 138.03 24.67 6.76 10.06 15.42 23.34 67.26 Boltz Avgs 0.898 204.62 76.89 31.41 137.72 139.27 81.08 54.22 141.32 139.51 24.21 6.62 9.78 13.42 20.94 67.88 差異 -4.0 -0.7 -0.6 -2.6 -0.9 -6.2 0.8 0.1 -3.5 0.5 -0.6 -1.1 0.8 -5.1 1.1
エクセルではRMS値は、実験値との差異に対してSTDEV関数で"=STDEV(##:%%)"で表示させることが出来ます。
今回は、ChemDrawでの予想値はRMS=7.0 ppm, ωB97X-D/6-31G*で求めた化学シフトはRMS=2.3 ppmと算出されました。
メーカーからレポートされている計算精度は2.0ppmですので、多少不満が残るものの悪くはない結果といえます。
NMR化学シフト計算結果の解析ではDP4, DP4+(実際には計算方法とのパッケージ)が良く利用されていますが、中身がとても複雑です。
私にはその使い方が良く判らないことに加え、解析にブラックスボックスが入らないほうが良いと考え、単純な統計解析にとどめています。
これを踏まえ、天然型のilludin SのNMR化学シフトを、Xeon E-5 1660 v2 (6コア、12スレッド、3.7 GHz)で計算してみました。
ステップ 計算内容 所要時間 配座数 メモ ① 配座解析 数秒 - 648配座を探索、48候補配座を発見 ②-1 HF/321G構造最適化 約30分 48 20 kJ/mol以内に12配座を発見 ②-2 ωB97X-Dによる構造最適化 約3時間 12 ③ ωB97X-Dによる化学シフト計算 約1時間 7
計算結果を同様にエクセルで解析しました。
illudin S 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 天然物実験値 200.6 76.2 30.8 135.1 138.4 74.9 55.0 141.4 136.0 24.7 6.0 8.7 14.2 15.8 69.0 Label 相対エネルギー
(kJ/mol)ボルツマン分布
(%)1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 M0004 -3.08 0.415 204.31 76.51 31.77 140.84 140.51 76.45 56.08 143.41 138.44 24.46 7.27 10.20 15.39 16.79 69.79 M0023 -1.76 0.244 204.17 76.17 32.79 138.86 135.15 82.57 53.67 141.69 137.05 24.81 6.92 10.04 12.69 17.54 70.41 M0007 0 0.12 205.43 76.62 32.71 138.07 135.44 80.63 53.63 139.06 138.13 24.80 6.86 9.72 13.76 19.68 70.92 M0009 1.36 0.069 204.78 77.09 31.36 139.75 139.89 77.53 56.00 141.64 139.88 24.24 7.17 9.88 16.22 15.77 69.57 M0001 1.49 0.066 203.86 76.67 30.68 133.25 144.17 76.79 56.62 146.27 137.57 23.92 6.93 9.42 14.99 15.42 69.56 M0020 2.14 0.051 204.13 76.41 31.40 141.25 139.75 75.17 55.90 144.14 136.13 24.60 7.33 10.38 15.65 15.87 69.58 M0003 3.01 0.036 204.54 76.22 32.43 139.63 137.67 76.91 56.58 142.44 136.52 24.60 7.05 10.10 13.64 17.96 70.26 Boltz Avgs -1.3 0.259 204.41 76.47 32.04 139.43 138.65 78.49 55.24 142.54 137.92 24.55 7.10 10.04 14.52 17.15 70.05 差異 -3.81 -0.27 -1.24 -4.33 -0.25 -3.59 -0.24 -1.14 -1.92 0.15 -1.10 -1.34 -0.32 -1.35 -1.05
今回はRMS=1.4 ppmと有意にスコアがよくなり、こちらの異性体のほうが可能性が高いといえます。
計算ではここまでです。
RMS値が5 ppmと大きくなれば、深く考えず除外できるのですが、今回の6-epi体では、RMS=2.3 ppmと計算のデータだけで除外することは危険です。
計算はあくまでも参考で、他のスペクトルデータと総合的に判断する必要があると思います。